AL Amyloidosis Background
Every protein has an assigned job, a schedule, and a freshly pressed lab coat. Antibodies clock in, fight infections, and clock out when their job is done. But what happens when one of those antibodies becomes unmoored—cutting corners, breaking rules, or acting outside its assigned role? That's when light chain AL amyloidosis can develop. Light chain AL amyloidosis is a rare and often underdiagnosed medical condition, characterized by multiorgan deposition of misfolded immunoglobulin light-chain amyloid fibrils that cause mechanical disruption, local oxidative stress, and eventual organ failure.¹ The most involved organs are the heart and kidneys, but the gastrointestinal tract, liver, and nerves may also be involved. The clinical presentation of AL amyloidosis often includes fatigue and unintentional weight loss but may also include additional symptoms specific to the organs involved.²⁻⁴
The clinical evaluation of AL amyloidosis begins with routine laboratory tests, including a complete blood count (CBC) with differential and a comprehensive metabolic panel (CMP). Additionally, serum protein electrophoresis with immunofixation will assess for the presence of a monoclonal paraprotein, while urine protein electrophoresis will quantify proteinuria and characterize urinary light chains. Serum free light chains should also be included in the diagnostic workup and, if present, confirmed by tissue biopsy. A fat pad biopsy is a specialized procedure where subcutaneous adipose tissue, typically from the abdomen, is collected for Congo red staining to confirm the presence of amyloid, but not the type of amyloid. The reported sensitivity is approximately 85% with multiple contributing factors that can affect the result: operator expertise, tissue handling, and biopsy site. A bone marrow biopsy and aspiration can confirm amyloid deposition. Once amyloidosis is identified, mass spectrometry-based proteomic analysis will assist in determining the type of amyloid present.1-4
Treatment Strategies for AL Amyloidosis
After a diagnosis is confirmed, the central questions are: how should we treat AL amyloidosis, and what is the likelihood of achieving a complete hematologic response (CHR)? A CHR in AL amyloidosis is defined as negative immunofixation in serum and urine and normalization of the involved serum free light chains (FLC). As a hematological response typically takes about 6-12 months as plasma cell dyscrasia responds to treatment. Organ function improvement on the other hand may take up to 24 months as amyloid deposits need to be reabsorbed for reversal of damage to be seen.
Before the ANDROMEDA trial, for eligible patients, the treatment paradigm often centered on proceeding to autologous stem cell transplant after high-dose melphalan conditioning to achieve a CHR, with results in studies from the 1990s to early 2000s ranging from 40% to 60%.6 High-dose melphalan followed by ASCT was compared to chemotherapy (melphalan and dexamethasone) by Gertz et al. showing a median progression free survival (PFS) (4 vs. 1.63 years) and median overall survival (not achieved vs. 6.53 years) to favor ASCT, while Jaccard et al did not show superiority between either arm.7,8 The comparison between efficacy and safety is also inconclusive as unexpected early mortality occurred in the ASCT arm and 26% of patients assigned to a transplant did not receive one.9 Treatment-related mortality was noted to be higher in patients with cardiac involvement, renal impairment, or advanced age in AL amyloidosis compared to multiple myeloma due to imminent risks like organ damage from existing amyloid fibrils and mortality related to cardiotoxic conditioning.6,9 Multiple trials have compared a dose reduction of melphalan conditioning from 200 mg/m2 to 100-140 mg/m2. However, variable CHR has been noted and showed a trend of decreased overall survival from approximately 10 years to 5 years with the dose reduction.10 Given that cardiac and renal involvement are present in approximately 75% and 60% of patients, respectively, and that the median age at diagnosis is approximately 65 years, a very small number of patients actually qualify for an ASCT with a good performance status and limited organ involvement, which resulted in an unmet need in the treatment strategies and role for ASCT in AL amyloidosis.
Treatment has evolved to target the underlying etiology of AL amyloidosis by utilizing multiple myeloma regimens, rarely Non-Hodgkin lymphoma regimens, to achieve organ and light-chain response, reduce clonal expansion, and improve patient tolerance prior to ASCT. Bortezomib-based regimens showed promising results after Kastritis et al. published a phase 3 trial that compared melphalan and dexamethasone (MDex) with bortezomib, melphalan, and dexamethasone (BMDex) in newly diagnosed AL amyloidosis. A total of 78% of patients who received BMDex, compared with 53% (P=0.001) of patients who received MDex, met the primary endpoint of a hematologic response at 3 months. Patients who achieved a cardiac response were still on the lower end and favored the BMDex (38% vs. 24%) along with a lower percentage of cardiac progression (BMDex 32% vs. MDex 15%, p=0.54). Renal response was similar in each arm. Overall, after a median of 25 months, survival did not differ significantly between the two groups.11 Pallladini et al. combined bortezomib with a different alkylator, cyclophosphamide, along with dexamethasone (CyBorD) and showed a hematologic response in 60% of patients, with 23% of those patients achieving a complete response (CR). The trial projected that 55% of patients would survive 5 years, with a median time to second therapy of 13 months.12 Smaller retrospective studies corroborated these results, demonstrating similar hematological responses, with Venner et al. demonstrating an overall hematologic response of 81.4% and an estimated 2-year PFS of 66.5% in 48 patients, while Mikhael et al. demonstrated a 94% overall response and 71% CHR in 16 patients in a smaller cohort.13-14
1a: Staging for AL Amyloidosis Based on Cardiac Involvement
| Prognostic Risk Variables | Value | Prognostic Variable Risk Score | Stage Based on Prognostic Variable Risk Scores |
| Troponin | Cardiac troponin T (cTnT): 0.025 μg/L and higher | 1 | Stage I (Risk Score = 0) |
| BNP | N-terminal pro-B-type natriuretic peptide (NT-ProBNP): 1800 mg/L or higher | 1 | Stage III (Risk Score = 2) |
The ANDROMEDA trial transformed treatment options for newly diagnosed AL amyloidosis patients who remain transplant ineligible. By adding daratumumab, a CD38-targeting monoclonal antibody to CyBorD, the median HCR at 6 months favored quadruplet-based therapy compared to CyBorD alone at 6 months (49.7% vs. 14%). Dara-CyBorD also showed a better cardiac (41.5% vs. 22.2%) and renal response (53% vs. 23.9%). As Dara-CyBorD established superiority as the preferred first-line for newly diagnosed AL amyloidosis, many of the patients with advanced cardiac or renal involvement were excluded: Stage IIIb-IV and estimated glomerular filtration rate less than 20 mL/min/1.73 m2 (see Figure 1c).15 Furthermore, Oubari et al. expanded the understanding of Dara-CyBorD in higher-risk populations. This retrospective study included patients with AL amyloidosis with Mayo stage IIIb and found that patients who received a daratumumab-based regimen reached the median overall survival at the 14.5-month mark of follow-up compared to 6.6 and 2.2 months for the bortezomib-based and other treatment groups, respectively.16 Yohannan et al. also retrospectively evaluated patients who received Dara-CyBorD compared to CyBorD and included advanced cardiac and renal AL amyloidosis. Patients eligible for cardiac response assessment achieved a higher cardiac CR or very good partial response (VGPR) with Dara-CyBorD (46.2% vs. 21.2% [P<0.001] and 76.6% vs. 43.6% [P <0.001]); patients eligible for renal response assessment achieved a higher renal VGPR or better in the Dara-CyBorD group (51.5% vs. 28.8% [P<0.001]).17 These findings indicate that for symptomatic patients with advanced cardiac involvement, Dara-CyBorD remains the best available treatment, provided cardiology clearance is obtained beforehand. Patients with Mayo stage IIIb with poor prognosis may start with monotherapy daratumumab and then re-evaluate the role or Dara-CyBorD.18 Similarly, patients with symptomatic renal AL amyloidosis, regardless of their eGFR, may also receive Dara-CyBorD after further discussion with nephrology.
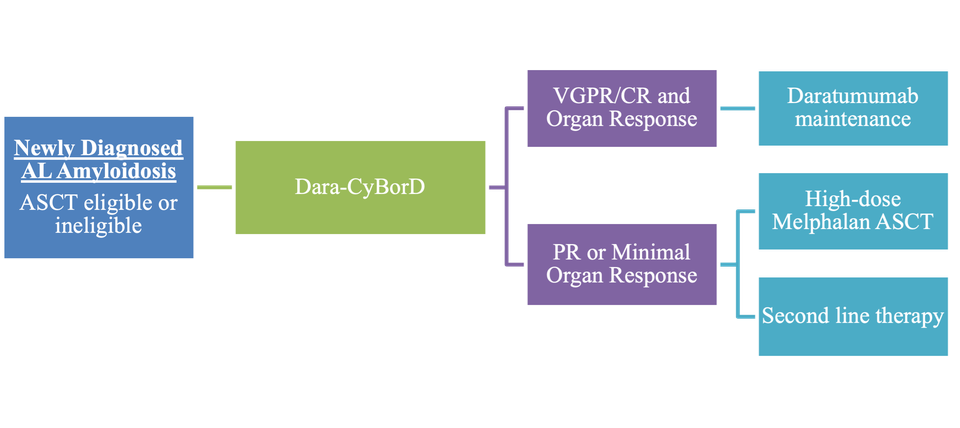
As induction therapy with Dara-CyBorD became the preferred front-line regimen for AL amyloidosis, the question remains regarding the place in therapy for high-dose melphalan ASCT. For patients who achieved a VGPR or better with organ response, a melphalan ASCT may potentially be deferred. If only a partial response (PR) or minimal organ response is achieved with Dara-CyBorD, then a high-dose melphalan ASCT may be considered, depending on cardiac and renal involvement.¹⁶ A single-center study led by Romera et al. demonstrated that even patients with cardiac amyloidosis could be safely treated with ASCT after achieving CHR from induction therapy. Cardiac response remained stable post-transplant with an NT-proBNP level of 511.5 pg/mL. Additionally, a median follow-up of 3.6 years revealed prolonged disease-free intervals, with only one death reported.¹⁶ It is crucial that patients with a possible indication for high-dose melphalan ASCT be evaluated at an experienced transplant center as superior outcomes have been noted with a reduction of early mortality.¹⁷ If the patient is ineligible for ASCT, alternative lines of therapy should be used.
References
-
Baker KR. Light Chain Amyloidosis: Epidemiology, Staging, and Prognostication. Methodist Debakey Cardiovasc J. 2022;18(2):27-35. Published 2022 Mar 14.
-
Al Saleh As, Sidiqi Mh, Muchtar E, Et Al. Outcomes Of Patients With Light Chain Amyloidosis Who Had Autologous Stem Cell Transplantation With 3 Or More Organs Involved. Biology Of Blood And Marrow Transplantation: Journal Of The American Society For Blood And Marrow Transplantation. 2019;25(8):1520-1525.
-
Ashutosh Wechalekar, Alberico Del Torto, Quarta C, Liedtke M. Al Amyloidosis For Cardiologists. Jacc: Cardiooncology. 2022;4(4):427-441.